What started in a scientist's dining room is now in tissue-agnostic combo trials.
In 1972, Janet Rowley sat at her dining room table and cut tiny chromosomes from photographs she had taken in her laboratory. One by one, she snipped out the small figures her children teasingly called paper dolls. She then carefully laid them out in 23 matching pairs—and warned her kids not to sneeze.
The physician-scientist had just mastered a new chromosome-staining technique in a year-long sabbatical at Oxford. But it was in the dining room of her Chicago home where she made the discovery that would dramatically alter the course of cancer research.
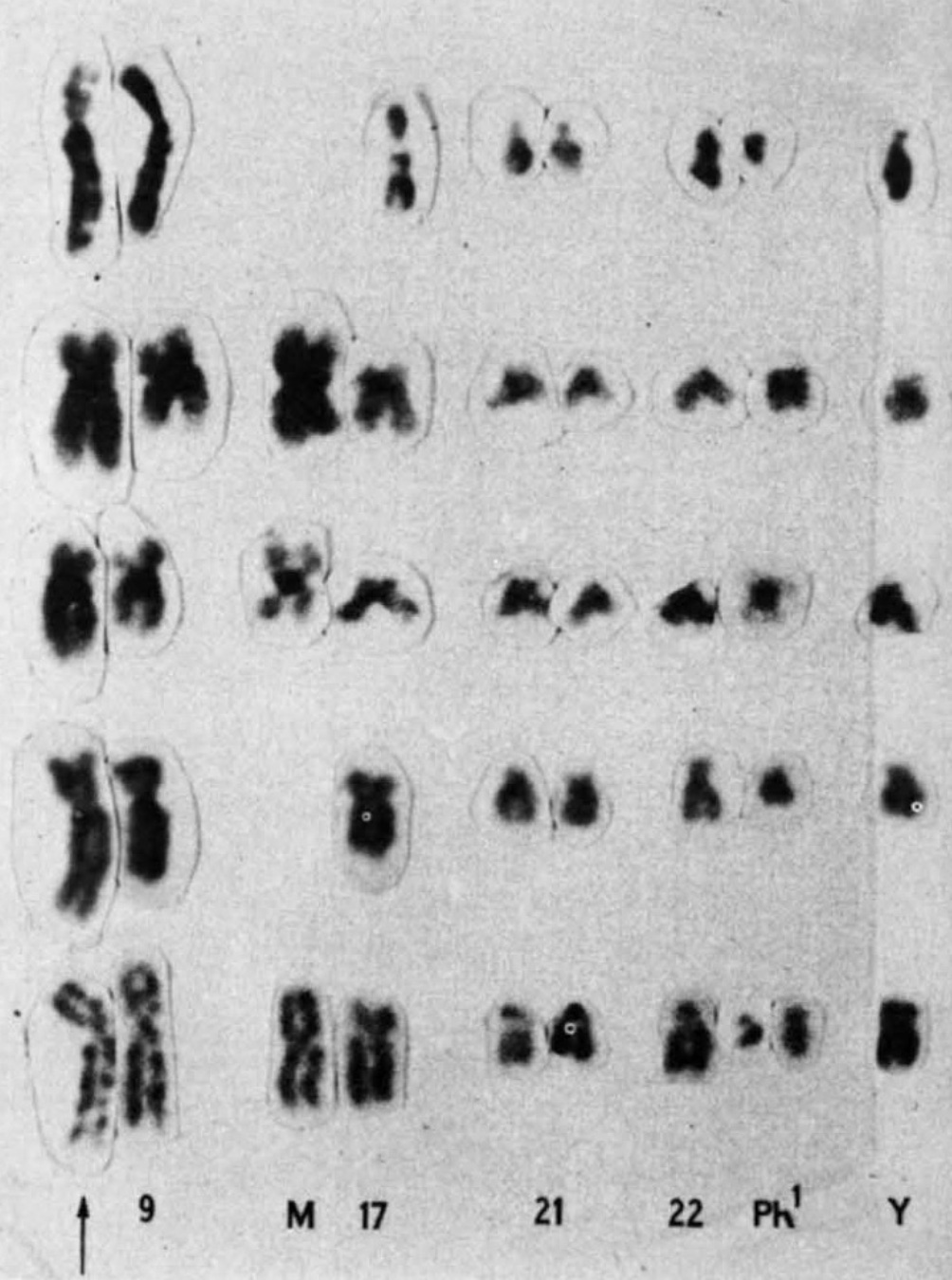
Rowley's 1973 partial karyotype showing the 9;22 translocation
Looking over the chromosomes of a patient with acute myeloid leukemia (AML), she realized that segments of chromosomes 8 and 21 had broken off and swapped places—a genetic trade called a translocation. She looked at the chromosomes of other AML patients and saw the same switch: the 8;21 translocation.
Later that same year, she saw another translocation, this time in patients with a different type of blood cancer, called chronic myelogenous leukemia (CML). Patients with CML were known to carry a puzzling abnormality in chromosome 22 that made it appear shorter than normal. The abnormality was called the Philadelphia chromosome after its discovery by two researchers in Philadelphia in 1959. But it wasn't until Rowley pored over her meticulously set dining table that it became clear why chromosome 22 was shorter—a chunk of it had broken off and traded places with a small section of chromosome 9, a 9;22 translocation.
Rowley had the first evidence that genetic abnormalities were the cause of cancer. She published her findings in 1973, with the CML translocation published in a single-author study in Nature. In the years that followed, she strongly advocated for the idea that the abnormalities were significant for cancer. But she was initially met with skepticism. At the time, many researchers considered chromosomal abnormalities to be a result of cancer, not the other way around. Rowley's findings were rejected from the prestigious New England Journal of Medicine. "I got sort of amused tolerance at the beginning," she said before her death in 2013.
The birth of targeted treatments
But the evidence mounted quickly. In 1977, Rowley and two of her colleagues at the University of Chicago identified another chromosomal translocation—15;17—that causes a rare blood cancer called acute promyelocytic leukemia. By 1990, over 70 translocations had been identified in cancers.
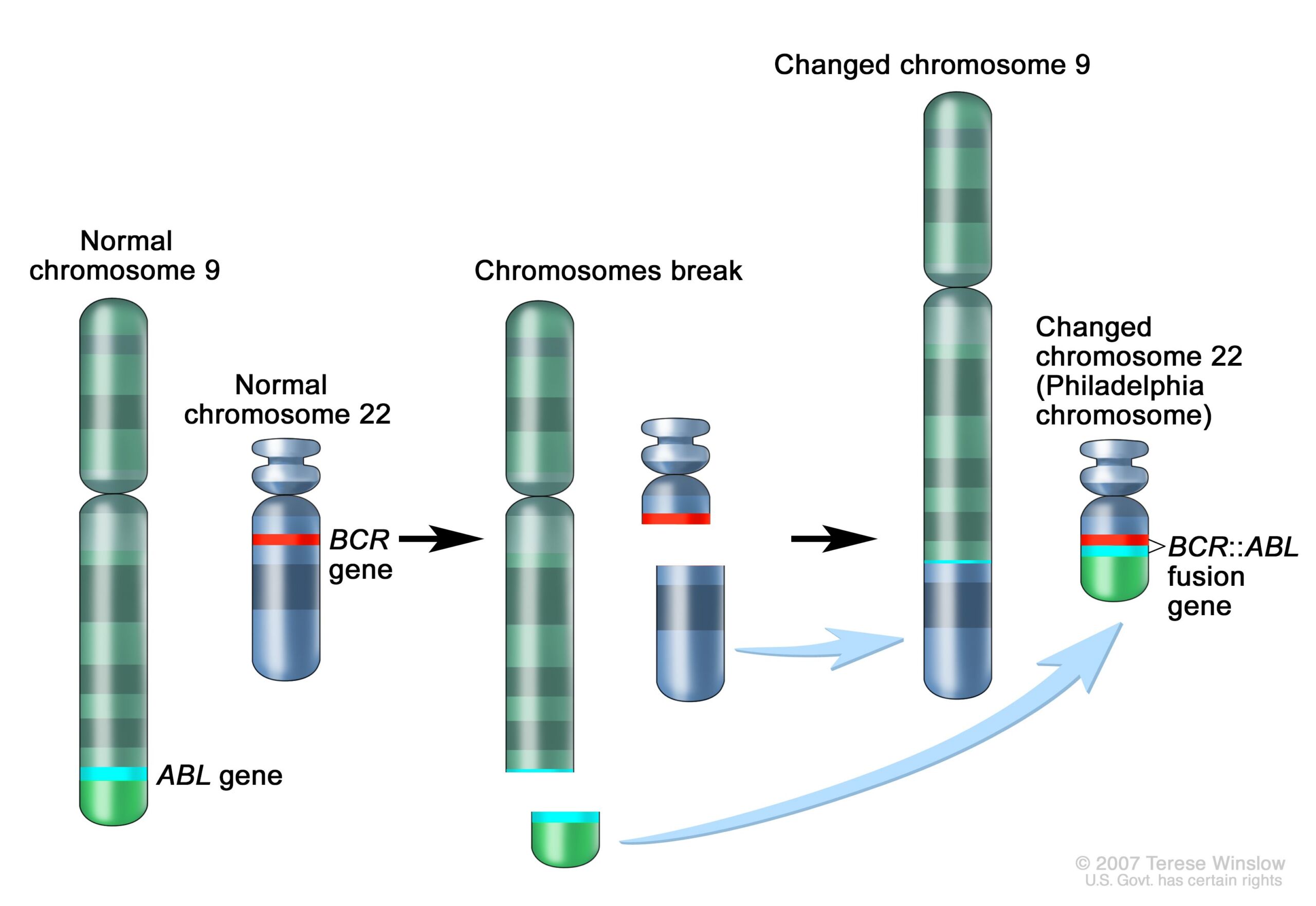
The significance mounted quickly as well. Following Rowley's discovery of the 9;22 translocation in CML, researchers figured out that the genetic swap creates a fusion of two genes. Part of the ABL gene normally found on chromosome 9 becomes attached to the BCR gene on chromosome 22, creating the cancer-driving BCR::ABL fusion gene on chromosome 22. This genetic merger codes for a signaling protein—a tyrosine kinase—that is permanently stuck in "active" mode. As such, it perpetually triggers signaling pathways that lead white blood cells to grow uncontrollably.
Schematic of the 9;22 translocation and the creation of the BCR::ABL fusion gene.
By the mid-1990s, researchers had developed a drug that blocks the BCR-ABL protein, a tyrosine kinase inhibitor (TKI) called imatinib. For patients in the chronic phase of CML—about 90 percent of CML patients—imatinib raised the 10-year survival rate from less than 50 percent to a little over 80 percent. Imatinib (sold as Gleevec or Glivec) earned approval from the Food and Drug Administration in 2001, marking the first approval for a cancer therapy targeting a known genetic alteration.
With imatinib's success, targeted cancer therapies—aka precision medicine—took off. By the early 2000s, there was widespread interest among researchers to precisely identify the genetic underpinnings of cancer. At the same time, the revolutionary development of next-generation genetic sequencing acted like jet fuel for the soaring field. The technology eased the identification of mutations and genetic abnormalities driving cancers. Sequencing is now considered standard care in the diagnosis, treatment, and management of many cancers.
The development of gene-targeting cancer therapies skyrocketed. Classes of TKIs, like imatinib, expanded particularly fast. There are now over 50 FDA-approved TKIs targeting a wide variety of cancers. For instance, the TKIs lapatinib, neratinib, tucatinib, and pyrotinib target human epidermal growth factor receptor 2 (HER2), which runs amok in some breast and gastric cancers. The TKI ruxolitinib targets Janus kinase 2, which is often mutated in the rare blood cancer myelofibrosis and the slow-growing blood cancer polycythemia vera. CML patients, meanwhile, now have five TKI therapies to choose from.
Evolutionary arms race
Unfortunately, though, the story doesn't end here. While targeted cancer therapies have made significant strides in cancer care—improving survival rates and buying patients priceless time—significant challenges remain. The sequencing advances that have sped the identification of mutations behind cancers have outpaced researchers' ability to interpret their importance in disease. This has led to a heap of VUSs, or variants of unknown significance, which require follow-up research to understand.
But researchers have also become painfully aware that cancers—especially advanced ones—can be heterogenous, with distinct groups of cancer cells driven by different mutations. Then there's the daunting and all-too-common problem of resistance. Cancer cells are adept at getting around targeted therapies, either through new mutations or pre-existing heterogeneity. Both can unceremoniously turn carefully researched precision therapies into duds.
Such is the case for some patients on imatinib; cancer cells have used several ways to get around the landmark targeted drug. Like many TKIs, imatinib works by locking into a pocket of the BCR-ABL protein, where it normally binds the energy-providing molecule ATP, or adenosine triphosphate. As a tyrosine kinase, BCR-ABL's role is to bind ATP and transfer one of its phosphates to a downstream signaling pathway, thus activating the pathway. With imatinib latched on, the ATP site is blocked, preventing BCR-ABL from binding ATP and activating the downstream pathways. But cancer cells can get around this with mutations in and around the ATP binding pocket that prevent imatinib from binding.
The evolving resistance has led to the creation of second- and third-generation TKIs. Newer drugs—nilotinib, dasatinib, bosutinib, and ponatinib—bind to the same ATP-binding pocket in BCR-ABL as imatinib, but they have firmer grips on the protein and are less affected by small changes in the binding pocket. An even newer drug, called asciminib, has raised hopes recently with a novel inhibition strategy.
Unlike the previous TKIs, asciminib does not bind to the BCR-ABL ATP-binding pocket, instead binding to a region of BCR-ABL called the myristoyl pocket. When it binds, asciminib locks the fusion protein into an inactive form, preventing it from activating downstream pathways. With its novel mechanism, it's expected to thwart all previous resistance mutations that have developed against the earlier drugs. Studies published in May suggest that asciminib is superior to other TKIs as a front-line drug and is effective in patients with a common resistance mutation.
One-two punch
While such new chemical tricks can bypass resistance mutations—at least for a time—researchers are increasingly looking to combination therapies, which could help prevent resistance from developing in the first place while boosting efficacy. Strategies include using two drugs that hit the same cancer target in different ways. The TKI therapies for CML are a good example; combination therapies could include an inhibitor that binds the ATP-binding pocket (imatinib) of the BCR-ABL fusion protein and another that binds the protein's myristoyl pocket (asciminib).
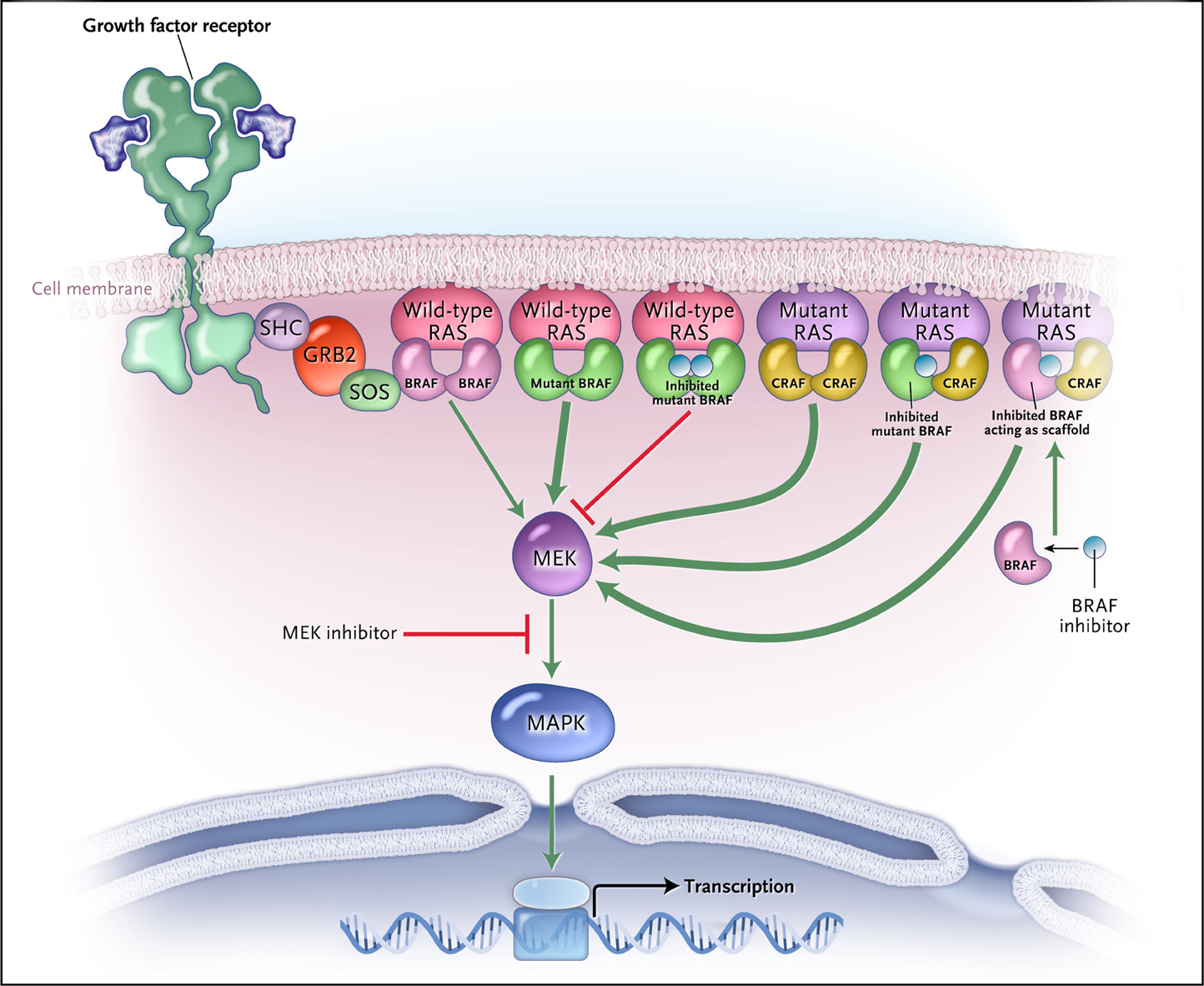
Another strategy for combination therapies is using multiple drugs to hit different components in the same—or a separate—signaling pathway. Researchers have recently found success using this strategy against cancers driven by BRAF-V600 mutations. BRAF is a serine/threonine kinase within a signaling pathway that, when overactivated, can lead to uncontrolled cell growth and a variety of cancers. Mutations in BRAF commonly lead to this overactivation, and the mutations often affect the valine at position 600 of the protein (hence V600).
Researchers have developed inhibitor drugs that can effectively shut down the activity of BRAF V600 mutants, including vemurafenib, encorafenib, and dabrafenib. But when given alone, BRAF inhibitors can paradoxically increase signaling in BRAF's pathway in some patients. It has been hypothesized that a sister signaling component to BRAF, called CRAF, may take its place after prompting by either inactivated BRAF or a mutated signaling protein upstream. In other patients, BRAF inhibitors work for a time, but then cancer cells go on to develop resistance by activating the components of the signaling pathway just downstream of BRAF, including one called MEK, also known as mitogen-activated protein kinase kinase.
RAS–RAF Signaling in Melanoma and models of how BRAF inhibition paradoxically leads to activation of the signaling pathway.
In recent years, researchers have used a combination of BRAF and MEK inhibitors, which avoids the paradoxical increased activity, decreases activity in the pathway, and staves off resistance. In a clinical trial published in 2015, a combination of the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib improved survival in patients with metastatic melanoma (a skin cancer) as compared to therapy with one BRAF inhibitor.
Genetic baskets
BRAF V600 is commonly found in melanoma, but in recent years, the dual therapy has drawn attention for successfully treating various cancers. In June 2022, the FDA granted accelerated approval for the use of dabrafenib-trametinib combination therapy against nearly any metastatic or inoperable solid tumors driven by BRAF V600 mutation. The approval opened the combination therapy to patients with cancers of the colon, rectum, lung, thyroid, ovary, and brain, among others. In March 2023, the FDA expanded access further for children with BRAF V600-driven low-grade glioma, a type of brain tumor.
The expansions fall into what's sometimes called a "tissue-agnostic" approach to treatment. That is, the treatment choices are directed by the genetics, not the location of the cancer.
The FDA's expanded approvals of the dabrafenib-trametinib combination therapy were based, in part, on data from studies run by the National Cancer Institute that are putting the tissue-agnostic approach to the test. Along with others, the institute is running "basket trials," in which cancer patients with various types of cancers are enrolled and assigned to studies and treatments based on the genetic and molecular features of their tumors. The trials are complicated to set up, but they can help identify the full potential of existing treatment options that might otherwise be overlooked.
For instance, the BRAF combination therapy was tested in the basket trial NCI-MATCH. "An important finding of our study was that several rare types of cancer responded to the [BRAF] drug combination, including ovarian cancers, which are difficult to treat," NCI-MATCH investigator Lyndsay Harris said in response to the 2022 FDA approval expansion. "Some of the patients had very long responses, lasting for years."
NCI-MATCH wrapped up in 2023 after collecting results from 39 basket substudies, which pointed to benefits of the tissue-agnostic approach. NCI has now moved on to next-generation trials, including ComboMATCH, which combine the basket concept with the successes of combination therapies.
"These next-generation trials build on the lessons from the past, incorporating the knowledge we've gained and building in the flexibility needed to rapidly implement new ideas and take advantage of the latest improvements in technologies," James H. Doroshow, NCI's director of the Division of Cancer Treatment and Diagnosis, said in an announcement.
Killer combinations
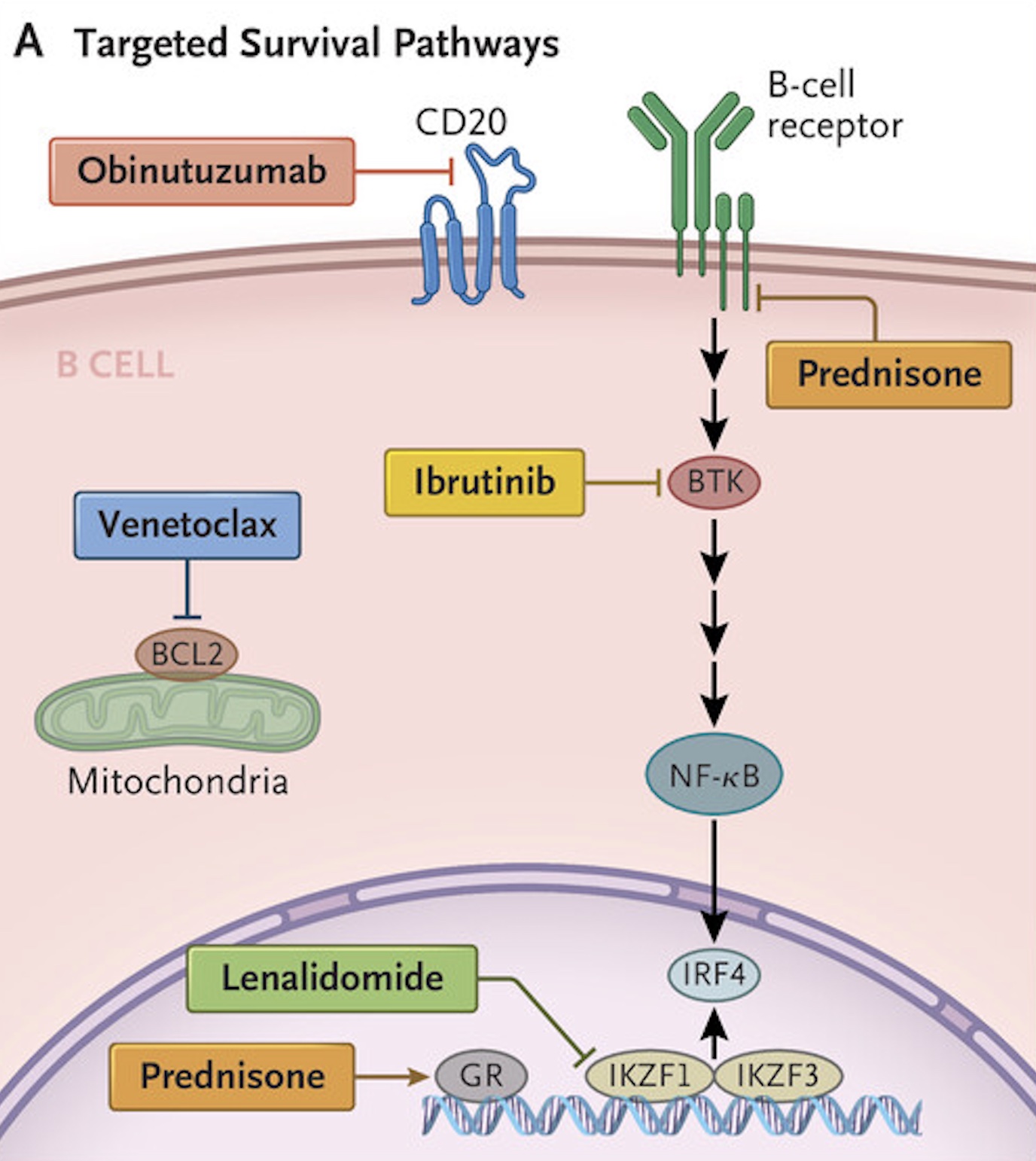
As the trials get underway, positive data on combination therapies continue to roll in. For instance, in June, NCI researchers published the results of a trial that combined five drugs to treat relapsed cases of diffuse large B-cell lymphoma (DLBCL), a notoriously heterogeneous and aggressive cancer. Genetic and functional studies had identified multiple druggable targets in cases of DLBCL that evade standard treatments, but using one targeted treatment at a time has shown little success. So researchers came up with the five-drug regimen that slams multiple cancer-driving pathways at once. The cocktail includes venetoclax, ibrutinib, prednisone, obinutuzumab, and lenalidomide, together nicknamed ViPOR.
Panel A shows the survival pathways targeted by the ViPOR (venetoclax, ibrutinib, prednisone,
The first drug, venetoclax, blocks a protein called BCL2, which in DLBCL would otherwise prevent cancer cells from undergoing self-directed cell death (apoptosis). Ibrutinib is a TKI that blocks Bruton’s tyrosine kinase, which is key to cancer cells' proliferation, and lenalidome leads to the targeted degradation of specific regulators, resulting in inhibited cell growth. Prednisone, meanwhile, is a corticosteroid known to spur cell death in cancers. The last drug, obinutuzumab, triggers innate immune responses that attack the cancerous B cells in DLBCL.
In the Phase 1b-2 trial, about half of patients responded to ViPOR, with 33 percent showing no detectable tumor DNA circulating in their bodies at the end of the therapy.
"Many of these patients who stopped responding to standard treatments would have otherwise died within a year, and now we have a good proportion who are still alive past two years, and some past four years," NCI researcher Christopher Melani, who co-led the study, said in a statement. "It’s gratifying to see these long-term remissions and potential cures in patients."
Next up
With expectations heightened for genetically matched combinations of existing drugs, researchers are still working on therapies with new chemical tricks that could be added to targeted cocktails in the future. For instance, there is excitement over new menin inhibitors to treat cases of acute myeloid leukemia (AML) with mutations in KMT2A, a histone methyltransferase that modifies the activity of DNA. Mutated forms of KMT2A found in AML bind to the scaffold protein menin, and the pair activate genes that lock the cell into a self-renewal cycle, leading to cancer. New menin inhibitors block the protein complex, preventing the aberrant gene activation. There are at least seven menin inhibitors in development, including one under review by the FDA.
Similarly, researchers are eagerly watching the development of drugs that target the metabolic enzymes isocitrate dehydrogenase 1 (IDH1) or 2 (IDH2), which are mutated in AML and gliomas. The mutated forms of IDH1 and IDH2 produce the metabolite 2-hydroxyglutarate, which accumulates in the cell and leads to DNA hypermethylation, changes in gene activation, and cell proliferation. A Phase 3 study published last year found that the IDH1/2 inhibitor vorasidenib can cross the blood-brain barrier and significantly slow the progression of malignant brain tumors.
Vorasidenib, the first new glioma drug to be developed in over 20 years, is now under review by the FDA. It is one of many other targeted therapies and new technical maneuvers in the pipeline to hone precision cancer treatments and get us closer to the ultimate goal of curative therapies. But it's also worth looking back on how far we've come. In March, Ingo Mellinghoff, lead investigator on the vorasidenib trial, acknowledged the steps it took to get to that breakthrough glioma treatment: preclinical studies, the genetic sequencing advances, and maybe even some tiny paper cutouts of chromosomes. The clinical success of vorasidenib "would not have been possible without all the research legwork and basic science," he said.
You can post now and register later.
If you have an account, sign in now to post with your account.
Note: Your post will require moderator approval before it will be visible.
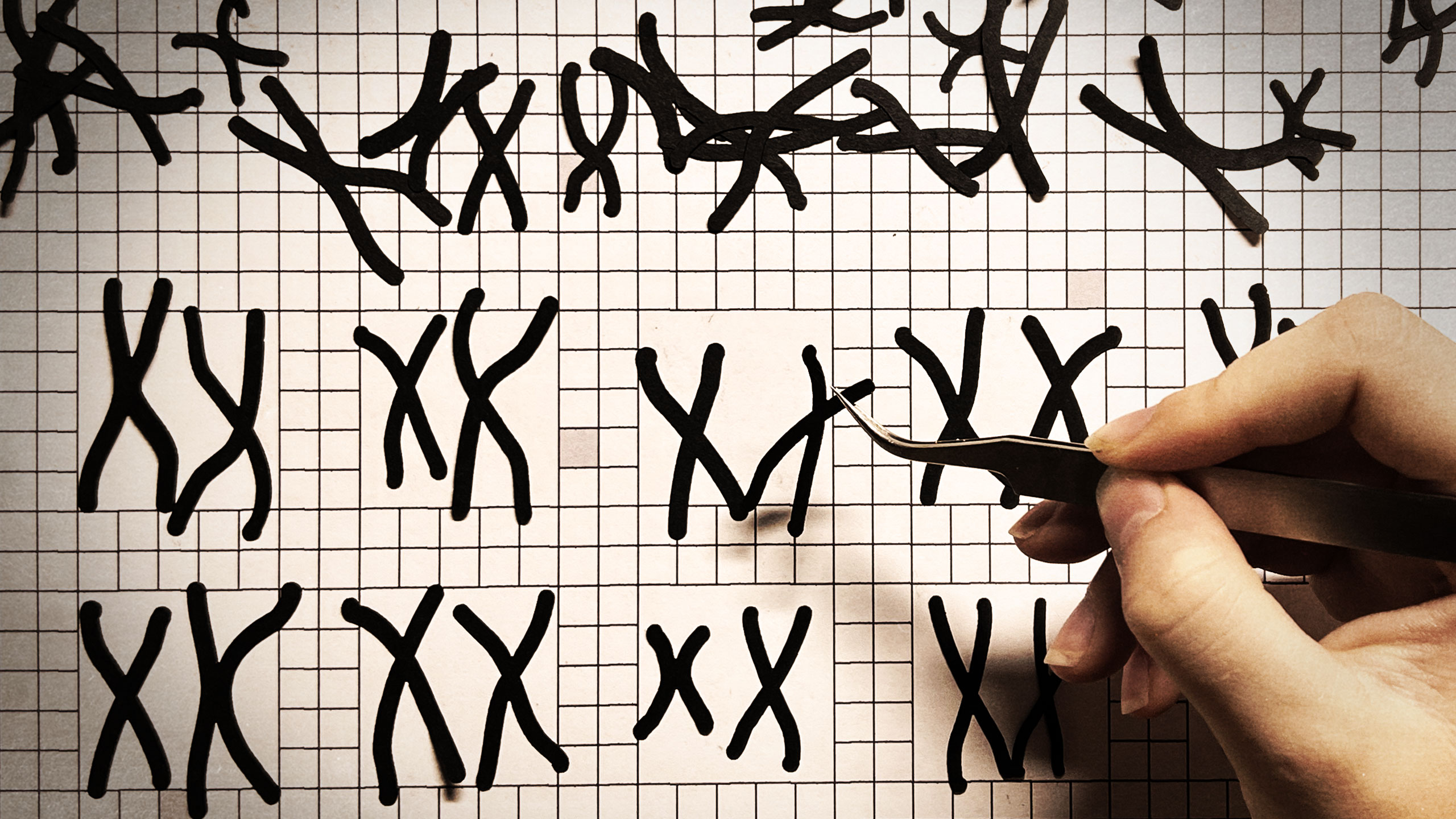
3175x175(CURRENT).thumb.jpg.b05acc060982b36f5891ba728e6d953c.jpg)

Recommended Comments
There are no comments to display.
Join the conversation
You can post now and register later. If you have an account, sign in now to post with your account.
Note: Your post will require moderator approval before it will be visible.