Blood stem cells are being engineered to protect them from lethal therapies.
Know your enemy, know yourself. It's a centuries-old strategy. But even in the present-day war against cancer, achieving it remains elusive. In many cases, cancer cells blend in with healthy ones. They bear no unique molecular markers or targets that we can aim clinical defenses at. That means any deadly strike on enemy cancer cells could result in casualties among healthy ones as well. The untenable toxicity of this artless warfare has led some researchers to rethink the ancient script—and flip it: know yourself, know your enemy.
In a set of clever and highly technical tricks, researchers are working on ways to precisely mark and shield healthy cells from chemical weapons, abandoning the effort to pick out enemy cancer cells specifically. By exploiting molecular markers common among many types of cells, researchers can safeguard healthy cells, leaving only the cancer cells in harm's way.
A drug or therapy that targets common markers would normally lay waste to cancerous and healthy cells alike. But that's not the case in this radical approach, which is first being used to treat blood cancers. For the strategy, researchers collect healthy blood stem cells and genetically engineer tiny, benign changes to a common molecular marker on them. Those tiny changes make the healthy cells essentially invisible to killer treatments. After the engineered cells are transplanted into a patient, clinicians can deploy the treatments. The cancerous cells that lack the genetic tweak are now easily killed by the drug or therapy, while the healthy engineered cells are left untouched.
The utility of the tactic doesn't end there. While cloaking healthy cells means researchers no longer need to know their specific enemy from healthy cells with precision, that imprecision opens vast possibilities. For one, it has the potential to create a virtually universal treatment for blood cancers. Whatever specific type of cancer is present, targeting a ubiquitous marker on blood cells and cloaking healthy cells will eradicate whatever strain of cancer is lurking among them.
"We no longer have to worry whether a tumor originated from a B cell or from a T cell or from a myeloid cell. It doesn't matter," transplantation immunologist Lukas Jeker of the University of Basel in Switzerland told Ars. "As long as it is a blood cell, we can use [the strategy], and that's why we think it's going to be almost universal."
For now, the results are still in the pre-clinical realm—mouse studies and Petri dishes—but researchers are swiftly marching toward clinical trials with compelling results in hand. Those trials using the strategy "are eagerly anticipated by the scientific and medical community," oncologist Miriam Kim of the Washington University School of Medicine wrote in Trends in Cancer in December. Those trials are now expected in the next few years.
A universal target
Last month, Jeker and his colleagues published the early results of their nearly universal strategy. It targets CD45, a molecular marker found only on blood cells, with the exceptions of red blood cells and platelets. It's called a pan-hematopoietic marker. In the study published in Nature, the researchers successfully used a potent treatment that kills anything with an unaltered CD45. The treatment is an antibody-drug conjugate (ADC). The antibody part of the ADC is a protein that can uniquely latch onto CD45 and, once attached, deliver a toxic payload bound to it, which contains an existing cell-killing drug. Normally, a CD45-targeting ADC would wipe out the entire hematopoietic cell population. But with healthy blood stem cells shielded by a tweak to their CD45, the ADC selectively kills off the unprotected and cancerous cells.
In mouse experiments, the strategy worked. "It was just unbelievably effective. I mean, the first time I saw the results, I didn't believe it. I thought something must be wrong," Jeker said.
First, the researchers injected mice with four different cancerous cell lines. After a single dose of the CD45-targeting ADC, all the rodents' tumors shrank rapidly. They then tested it out with a cancer patient's cells. The patient had a type of blood cancer called AML, Acute Myeloid Leukemia, which is a kind of blood cancer that originates in the bone marrow. The researchers transplanted the patient's AML cells into mice that had already gotten a transplant of healthy CD45-shielded blood stem cells. They then treated the mice with their CD45-targeting ADC.
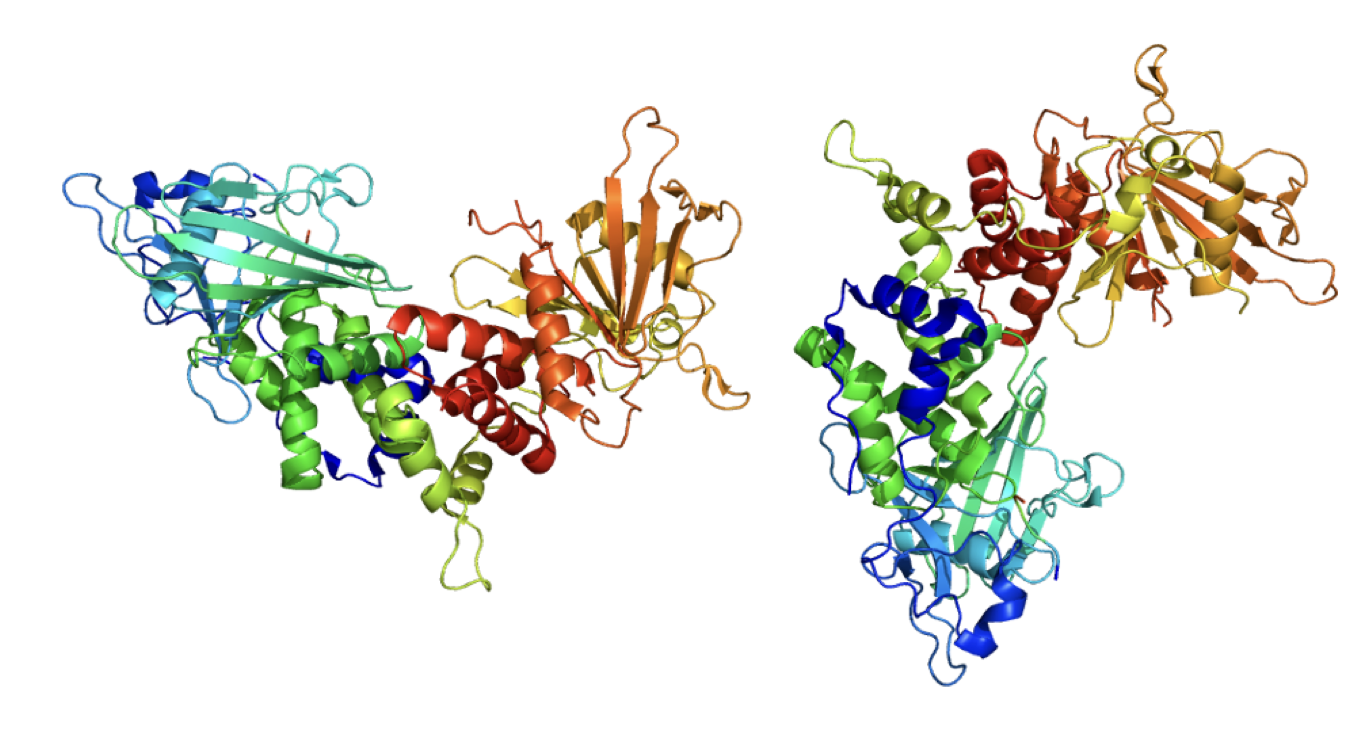
The CD45 protein, present on almost all blood cells.
"The results were really just black or white. It's just like, it just works," Jeker said. The CD45-shielded cells were protected. The researchers saw no evidence of them being killed. Meanwhile, the patient's AML cells were eradicated. "So we really get the dream result," Jeker said. "We have the complete protection, and we get complete elimination of the disease, which is what we want, ultimately."
How the shielding works
Of course, getting to those dream results took a lot of work. Jeker's study builds on results published last year by two other research groups, one led by researchers at the University of Pennsylvania and the other at Boston Children's Hospital and Harvard. In both cases, researchers used new gene-editing tools for a technique called "base editing" on blood cell markers. Base editing, which uses CRISPR/Cas9 machinery, allows researchers to precisely edit a single base—a nucleotide building block of DNA—in a gene. Unlike other editing techniques, this doesn't require double-stranded breaks of the genetic material, which can be toxic to cells. It's a precise and less toxic editing method.
The goal of the single base edit is to change the molecular marker just enough to keep an antibody from recognizing it. Mouse-generated antibodies are critical tools in the fight against cancer. Researchers train the antibodies to precisely recognize and bind specific molecules on the outside of cells. Once bound, antibodies can deliver toxic payloads, neutralize the function of a target, or recruit immune responses to attack and kill bound cells, among other useful tasks. But without molecular markers that are specific only to cancer cells, antibodies can be toxic to healthy cells. That's where the base editing comes in.
The goal is to "find a mutation that avoids the binding but at the same time preserve the functionality" of the marker, said gene therapy researcher Pietro Genovese of Harvard and Boston Children's Hospital. Genovese's group was behind one of the papers last year that provided a springboard for the development of the strategy. In the paper published in Nature, Genovese and his team base edited three markers present at high levels on AML cells: CD123, CD135, and CD117. Researchers already had antibodies against all these markers, which are used in the development of AML immunotherapies. So they just needed to find small tweaks that prevent the antibodies from binding in order to shield healthy cells.
Finding the right spot to base edit is another "trick we exploit," Genovese explained. Because researchers use mice to generate therapeutic antibodies, the result is a mouse anti-human antibody. Mice also have the blood cell markers researchers are targeting here, such as CD45, CD123, and so on. Parts of these markers are conserved—that is, the same—between humans and mice.
But the antibody "will never mount an antibody response against a self-antigen," Geneovese said, meaning that any parts that are mouse-specific or conserved between mice and humans likely won't be targeted by the mouse anti-human antibody. That leaves non-conserved, human-specific places for the antibodies to bind on the markers. If the area where the antibody binds is not conserved, it's likely not important for the marker's function, which could play a role in cell signaling or another activity.
Thus, targeting the area where the antibody recognizes the marker—an area called an epitope—takes advantage of known biases. Any changes in the epitope are likely to thwart both the antibody's binding and preserve the marker's function. Of course, all of those things still require testing to confirm and ensure the safety of the approach.
For the epitope binding, we're looking for a "minimal" genetic change to shield the cells, Jeker explained. "The minimal change is that we really change, ideally, just one amino acid from the hundreds of amino acids that are found in a protein," Jeker said of the base-editing technique. "We went through quite some lengths to identify, first, which antibody is the best one, which region is the best one, which exchange or which amino acid is the best position, and then also which substitution per position is the best one, because each position you can exchange against different amino acids." Jeker and his team further characterized the thermal stability, structural changes, and biophysical characteristics for potential base edits. Since this edit will persist in healthy stem cells, it's critical that this shielding edit is completely benign.
Clinical attack plans
In future patients, the approach would combine all the different moving parts. Currently, blood stem cell transplants (hematopoietic stem cell transplantation, or HSCTs) are used to try to cure a variety of blood cancers. Typically, the treatment starts with an unspecific chemotherapy that kills cancer cells as well as healthy cells, followed by a transplant of healthy stem cells, sometimes from the patient or sometimes from a cancer-free, healthy donor.
The healthy, transplanted stem cells go on to replenish the population of blood cells. In some cases, such as in leukemia patients, white blood cells from a donor transplant can attack lingering cancer cells from the patient, an effect called graft-vs-tumor or graft-vs-leukemia. This indiscriminate killing-then-replenishing approach can work for some patients. But aggressive cancers often return. Not all of the cancer cells may be killed by the chemotherapy; some may develop resistance to the drug treatment, and sometimes, the graft-versus-tumor effect doesn't work at all.
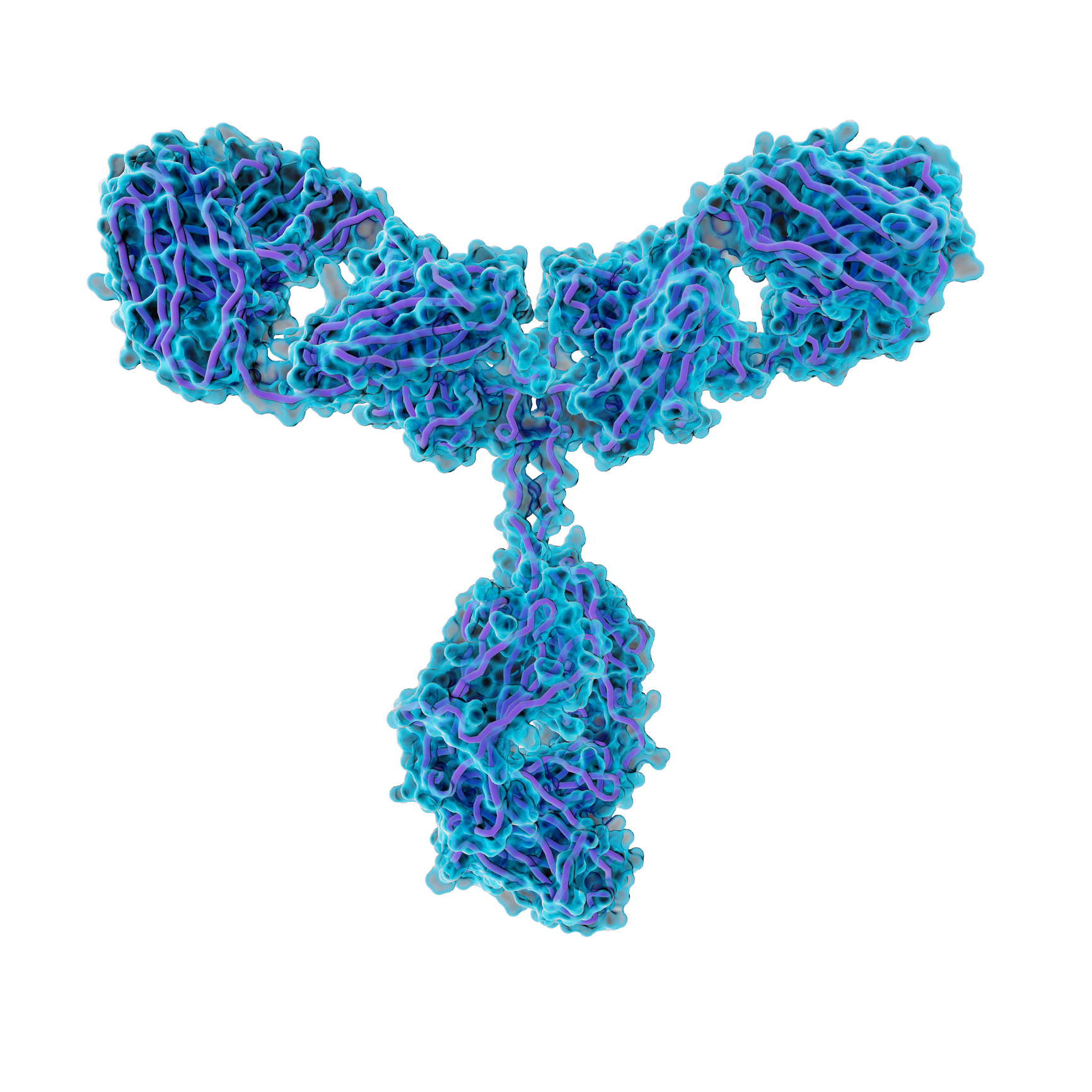
Antibody molecules, like the one shown here, can be linked to toxins and delivered to cells that carry a protein that the antibody recognizes.
With the new base-editing cloaking strategy, the risk of a relapse could be dashed. The treatment could start with chemotherapy, but the subsequent HSCT would introduce the shielded stem cells with base-edited CD45s or CD123s, for example. Once the transplanted stem cells are established, clinicians could treat the patients with targeted cancer-killing therapies such as anti-CD45 ADCs. The treatment would then kill off any lingering cancer cells and cells leftover from the original blood cell population.
If this initial strategy works in further pre-clinical testing as well as in early clinical trials in patients, Jeker said researchers could move to a next-level approach. That would involve skipping the toxic chemotherapy step in the beginning, and using the targeted therapies, such as anti-CD45 ADCs, instead. This approach could be safer for patients because it would selectively kill only blood cells, efficiently depleting the population before a transplant and potentially reducing toxicity.
Killer weapons
Further, the approach could also use different killing strategies. Jeker's group started with an antibody linked to an existing cancer drug. But Genovese's group used another genetic engineering tool called CAR T cells. In this strategy, researchers alter T cells—which are white blood cells involved in immune responses—to carry highly engineered receptors known as chimeric antigen receptors, or CARs. The CARS reach out from the T cell's surface and have a receptor made from components of synthetic antibodies.
These synthetic antibody domains on the CAR T cells work similarly to the antibody therapies described earlier; they can be trained to bind to specific molecular markers. Those molecular markers can be base-edited to prevent binding on healthy cells. After such shielding, the CAR T cells can target unaltered cancerous cells by binding to the targeted molecular marker. Once bound, the T cells directly kill the cancerous cell and recruit immune responses.
Genovese and his colleagues used CAR T cells to target two markers, CD123 and CD135 (aka FLT3). In this case, the researchers weren't working to target a universal blood marker like CD45 but instead markers that are known to be present at high levels in AML cells. "I would prefer ... to focus and concentrate the therapy only on the cells that you really need to kill in order to achieve the proof of concept [and] to provide therapeutic benefit" for early patients, he said.
So far, this approach has presented impressively positive results in mice. In mice that were transplanted with shielded (CD123 and CD135) stem cells and also patient-derived AML cells, treatments with anti-CD123 and anti-CD135 CAR T cell treatments worked—the shielded stem cells were protected, while the AML cells were eradicated.
In the other study published last year by the group at Penn, hematologist Saar Gill and immunologist and oncologist Carl June added yet another level of complexity to the approach. They, like Jeker, went for a universal marker, base-editing CD45 to shield healthy blood stem cells. They also used an anti-CD45 CAR T cells to kill off cancerous cells. The trouble is that T cells also contain CD45. For the approach to work—and keep the CAR T cells from simply massacring each other (called fratricide)—the researchers had to shield the CD45 on the T cells and those on the healthy stem cells.
As in the other studies, the strategy worked in mouse models. The base-edited shielding of CD45 didn't appear to alter cell functions and protected both the T cells and the stem cells from the anti-CD45 CAR T cells. The anti-CD45 CAR T cells, meanwhile, efficiently killed off patient-derived AML cells, B cell lymphoma cells, and acute T cell leukemia cells. The results were published in Science Translational Medicine.
"We’re very determined to get this into a clinical trial," Gill told the NIH last year. "It’s the only way we’ll know if it’s going to work."
March to trials
Genovese and Jeker both think they can have their strategies in clinical trials in a few years—less than five, Jeker said. The variations between their cloaking strategies have their own pros and cons, which will likely play out in clinical trial designs and the data. For now, Genovese is focusing on markers found at high levels in cancer cells, hoping they lead to a potent and highly effective therapy, while Jeker is optimistic about the universal approach with CD45.
While the results have been promising so far, most of the work has only been tested in mice.
Not all cancer cells contain high levels of CD45, Jeker noted—some are so low that they've been considered CD45-negative. But Jeker's group tested its anti-CD45 ADC on such a cell line in its experiments, finding that the cells were almost completely killed off in transplanted mice. Though tumor cells later relapsed in those mice, Jeker was still pleased with the result. "I'm actually very happy that even if they express very low levels, they still initially responded," he said.
The different killing strategies also present a choice for future development. Immunotherapies, like the ADC that Jeker's group used, have the advantage of combining off-the-shelf products (an antibody and an existing cancer drug) that are known to be effective. CAR T cell therapies, meanwhile, have the benefit of being a living cell line. The engineered T cells can persist in patients and have immunological memory to prevent relapses. But they also add a second layer of genetically edited cells to the strategy, making a regulatory review of safety and efficacy more complicated. Because the two components work together, it will be difficult to test their safety and efficacy separately.
"The real challenge will be to explain all this complex stuff to the regulatory agency and get the proper feedback to design a trial," Genovese said.
He also worries about the ethical considerations of their dual cell therapy approach. To ensure the strategy is not toxic to humans, we'll want to start with low doses in early clinical trials. But at low doses, a patient must go through a complex, risky treatment with low chances of efficacy—tipping the risk-benefit analysis against the trial. So the challenge, he says, is finding a way of starting with low doses and then scaling up to reach "a level in which you can really get efficacy."
Both Genovese and Jeker note that having two genetically engineered products in a clinical trial is not unprecedented. Currently, Vor Bio, a Boston-based biotechnology company, has a shielding strategy in early-stage clinical trials. It involves using an anti-CD33 ADC after fully deleting CD33 from blood stem cells in patients with relapsed AML. The company recently began moving forward with testing anti-CD33 CAR T Cells. (Both Genovese and Gill are clinical advisors to Vor).
If all goes well, Genovese and Jeker see the potential for the shielding strategy to move beyond AML to other blood cancers that could be treated with stem cell transplants—leukemias, lymphomas, severe myelodysplastic syndromes, and myeloproliferative neoplasms like high-risk polycythemia vera. Further, the treatment could move to other types of cancers, such as melanoma and colorectal cancer, auto-immune diseases, or even HIV infections.
"Of course, it's very complicated," Genovese said of the strategy. "But at the same time, I also feel pretty confident that it is feasible." Gene therapies and technologies such as these are already making their way into clinic, and "they are really effective therapies the moment some of them are becoming approved," he said.
You can post now and register later.
If you have an account, sign in now to post with your account.
Note: Your post will require moderator approval before it will be visible.

3175x175(CURRENT).thumb.jpg.b05acc060982b36f5891ba728e6d953c.jpg)

Recommended Comments
There are no comments to display.
Join the conversation
You can post now and register later. If you have an account, sign in now to post with your account.
Note: Your post will require moderator approval before it will be visible.